体外毒性测试
关于医疗器械测试,这里简单地分为两大类:体外测试及体内测试。首先来看体外测试,举几个例子:包括细胞毒性,溶血,遗传毒性……
什么叫做体外毒性测试呢?就是不需要动物做的测试。像细胞毒性,我们用小鼠型纤维细胞;像血液相容性测试、溶血测试,我们用兔子血或人血;遗传毒性测试,通常我们用细菌和小鼠淋巴瘤细胞。之所以选择用细胞或体外测试,主要为了减少动物使用,考虑动物福利的原因。国内也越发注重动物保护动物福利,以后可能会立法。新加坡、法国、欧洲都有比较激进的动物保护者采取了针对实验动物的保护行动。
因此,现在越来越多研发体外测试或替代测试方法。因后者是比较灵敏的筛选性测试。比如细胞毒性测试大致7天能得到结果,对后续研发或进一步测试很有指向性。但体外测试无法完全代表体内环境的情况。人体内环境有内环境稳态,包括免疫系统,酸碱平衡,耐受性容错性更好,因此,不能完全代替体内环境。因此需要做体内测试,选择动物模型,更接近人体解剖结构和生化条件。
研发皮肤类,选择家兔做模型,因其皮肤更接近人体皮肤。研发心血管类产品,倾向于选择猪,因其心脏结构最接近人体。因此,当我们希望在更深层次研究和探寻器械功能是否合用的情况下,先做动物测试。当然,要考虑动物福利322原则,尽量减少动物使用,考虑动物的伦理问题,人道和科学地对待动物,不能滥用、虐待动物,对实验进行详细计划,尽可能多地获取实验指标。避免重复、无益实验。
...
生物相容性影响因素
进一步介绍生物材料测试。哪些材料和成分与人体相容性相关,即与人体是否相容。以前更多讲的是被动相容性,即对人和动物不产生危害。现在有个新的概念,是主动的生物相容性。即不仅不对人体产生危害,而且还能对人体产生有益作用。因此,目前新的医疗器械的研发对材料提出了新的要求,比如还能促进骨组织愈合,诱导骨组织的生长。
在生物材料的生产过程中,以下因素会影响生物相容性:原材料,直接相关;生产加工工艺过程中的添加剂、污染和残留物,比如着色剂和染色剂;脱膜、溶解原材料再注塑的过程中引起的污染;残留物主要指灭菌和消毒过程中的残留物;滤出物指自然条件下和人体长期接触过程中产品释放的物质;降解产物,指人体内的植入物长期接触中产生的降解产物;以及其他终末产品与包装材料等接触带来的组分。产品设计中的物理和化学特性,都会影响生物相容性问题。
...
测试流程
进行材料的生物学评价过程,第一步是做化学表征测试。比如,医学级材料是否经过USB classic证书,ISO10993测试等。化学测试仅能测量材料本身是否含有有毒有害物质,还需进一步做动物测试来证实安全性,需按照ISO10993-1部分的附录来筛选动物测试,考虑材料在人体内接触的时间和部位,以及临床应用和已有文献数据来确定测试方法和计划。
所有有源医疗器械和无源医疗器械的安全性,都应按照以下流程图进行测试。
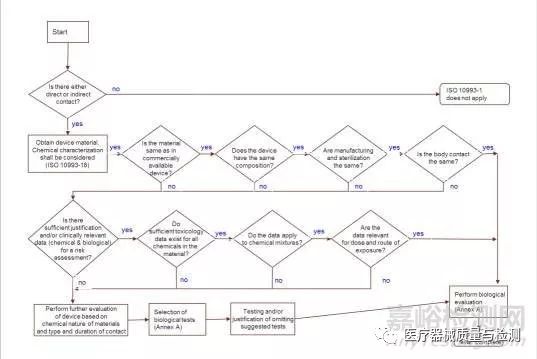
IVD产品因和人体没有直接接触,不适用ISO10993原则。首先,先看产品与人体有无直接接触,比如膏药,纱布,康复类器械,牙组织体,心血管支架等。间接接触就包括输液器,吸氧雾化器导管,通过管路进入人体内部,经由液体和气体接触人体。如此,判定是否需要按照此流程来进行测试。首先考虑化学表征对人体是安全的。有机和无机的化学成分都需要进行测试。通常原材料供应商会提供化学表征测试报告。当采用原材料供应商提供的材料生产医疗器械,也需要提供化学表征测试报告。接下来,考虑材料在上市产品中已有使用。
另外,器械中组分是否使用了其他材料。生产加工和灭菌过程是否对安全性产生问题。产品与已上市产品相比,与人体接触和使用过程是一致的。如果所有流程都通过认定。则可进行下一步生物学评价。当其中出现不符合某一流程的情况时,需要用文献数据,测试数据来支持测试报告。最终,根据测试报告,分析、解释,对产品的安全性进行阐述,是通过测试还是返回再次研究。
ISO10993中提出了八项原则。围绕医疗器械的评价流程图,要求有紧密的计划,同时强调试验并非唯一结果,只代表评价医疗器械材料安全性的一部分,需要综合判定。从实验设计到发生变化后,这8项原则都需要考虑,一直伴随研发器械和上市后过程。比如,患者和医生产生临床需求后,研发和市场人员考虑是否要研发这一产品,在研发和改进过程中均需考虑生物相容性。当所有研发过程完成后,再考虑是否满足法律要求。再进一步进行临床研究,上市后是否有不良事件,竞争对手是否有不良事件等。再对自身产品进行优化和改进。
通过与人体的接触,医疗器械可分为三类,一类是表面接触类器械,如纱布等。外部接触类器械,如内窥镜,插管,血液透析类设备等。高值耗材类器械,比如支架等植入性器械。按照接触时间进行分类,包括少于24小时,大于24小时,小于30天;大于30天,风险程度依次提高。随着时间接触越长,需要考虑的风险越多,要求的测试标准越高。
...
化学表征测试和生物学评价
生物相容性的评价过程就是考虑化学表征和测试的过程。
材料标准考虑化学测试,是按照ISO10993-18部分,按照与病人接触的时间和部位进行。同时考虑是否需要进一步的生物学评价。目前国内医疗器械测试和评价,此部分是热门且重要话题。举例:心血管支架已出现药物洗脱支架,可降解支架,是高风险类产品,长期接触,基本需要做全套的生物相容性测试。国外大致需要100万人民币,周期很长,需进行植入性实验等。国内更多接受化学表征测试和生物学评价。因器械设计已经非常成熟,主要风险来源于材料,考虑安全性便主要考虑其材料的安全性。
国外认为,应用化学方法检测材料的化学特性来判定安全性。精度达到ppM,或ppp。百万级甚至更高级别,灵敏度和精度都很高,以此免除进一步的生物学测试,包括动物学测试等。但国内的标准要求仍比较保守。现阶段,对国内企业的要求太高,因为找不到能独立完成全部化学表征测试的企业和毒理学评价专家。因此,目前更多的事依靠动物测试来做生物学评价。
关于和人体直接接触部位的反应,是化学表征测试无法代替的。植入过后反应需要用植入测试来评价。做生物学评价过程中,并不都需要做动物测试。比如,目前最常见的椎间融合器,选择的是peak材料,还有骨科的骨钉、骨板等是合金材料,有很长使用历史,原材料厂家有很全面的实验数据。生产厂家只是选择其进行制造,因此不需要进行额外的测试。可以通过寻找测试数据,而不是重新做实验,来证明其生物安全性。
但若用peak材料拿来做关节、牙组织体等新材料。通过机械切割达不到铸件要求,需要熔融和重塑,对化学有重大改变,则不能使用之前的化学报告,需要做额外的动物实验。
...
ISO10993生物学相容性测试
介绍ISO10993-1及整个标准的重要章节中的生物学测试,体外测试,包括细胞毒性测试,致敏测试(-5章节中)和皮肤刺激性测试(-10章节中)。急性全身毒性测试(-11章节中)。
大部分生物学材料都归于二类中,比较安全。因此主要做此三项测试:细胞毒性,致敏,皮肤刺激性测试。基本所有医疗器械均需做次三项测试。此外,还有急性系统毒性测试,亚急、亚慢性系统毒性测试。还有与人体接触时间更长者,需做遗传毒性测试。植入测试,血液相容性测试,包括溶血测试,血栓形成性测试、补体测试、凝血性测试(-4章节中)。以及慢性毒性测试、致癌性测试、生物降解性测试、生殖毒性测试,免疫毒性测试,毒代动力学测试等。
大多数研发企业不会考虑到材料改变,而是设计上的改变,因此较少涉及材料重新做测试项目的情况。现在较热的三个测试是毒代动力学测试,免疫毒性测试,生物降解性测试。因更多企业在做新的研发尝试,使用更多动物源性材料和可降解性材料。
如何进行测试,或与测试机构沟通测试需求。首先,需要制备浸提液。制备浸提溶液,将材料浸泡在其中。分为极性(生理盐水,细胞毒性测试是特例,用培养基),非极性浸提液(植物油,包括酒精,植物油和菜籽油),额外的浸提液(带血清的培养基、DMSO、水和酒精的混合物)。如何选择浸提液,根据相似相溶原理,选择浸提液就是根据产品所含材料的化学特性选择。让企业自行筛选的原因是,产品在某些浸提液中发生溶解,改变了产品特性,不符合测试要求。筛选浸提比例是送检时非常重要的环节。
所有测试方法一致的前提下,浸提比例会影响测试结果。针对产品的浸提比例,可选择厚度小于0.5mm时,选择6cm2/ml,大多数选择0.5mm厚度以上,用3cm2/ml。弹性体孔隙结构比较高时,选择1.25,不规则体选择0.2g/ml。吸水性物体密度很低,选择0.1g/ml。还有温度和时间的选择,细胞毒性的测试比较特异,应选择37度,72小时。还有50度,72小时。70度,24小时。121度,1小时。
由企业根据产品特性和临床使用要求自行选择。选择适宜温度和时间进行。较少选择37度,因为美国FDA不认为37度为严苛条件。而动物材料,如含有胶原蛋白者,可以选择37度。
任何与人体接触的,均需做测试。可以选择完整器械或部分器械做测试。举个实际惨痛案例。一款带药支架,准备做CE Mark测试,未选择完整器械。认为药物是安全的,有相应说明,只选择不带药器械做测试。通过公告机构的评审,在欧洲注册成功。2年以后拓展后做美国FDA测试,因未拿完整材料,需重新做测试,花费100万。建议一定选择上市的完整样品送检。同时考虑市场策略,资金较紧张时,国内检测所的要求和费用比较低。但产品需要进入国际市场,则一定要选一个严格的第三方,做一套完整的测试报告,否则费时费钱。另外,在切片的问题上,也有企业惨痛经验。因在产品制造过程中,管腔横切暴露未做涂层的部分,也会影响测试结果。
具体测试有细胞毒性测试,快速筛选性测试,选择用哺乳动物细胞,最常用的是小鼠神经细胞L929,方法有浸提法,直接接触法,非直接接触法(琼脂扩散法、滤膜扩散法)。评价方法包括在显微镜下看生长、形态、代谢情况。材料与人体血液有接触时,做细胞毒性测试。












